
Ostatnia aktualizacja 28 lipca 2022
Amerykańska Agencja ds. Żywności i Leków poinformowała firmę Sanofi, że planowane przez nią badanie rzeczywistego zastosowania (AUT), mające na celu wsparcie zamiany leku Cialis® (tadalafil) z produktu leczniczego wydawanego na receptę na produkt leczniczy wydawany bez recepty
zostało wstrzymane.
Cialis jest lekiem dostępnym wyłącznie na receptę, w postaci tabletek stosowanych w leczeniu zaburzeń erekcji (ED), oznak i objawów łagodnego przerostu gruczołu krokowego (BPH) oraz zarówno ED, jak i oznak i objawów BPH. Cialis jest jedynym lekiem z grupy inhibitorów PDE-5, który oferuje mężczyznom wybór w zakresie leczenia zaburzeń erekcji – Cialis do stosowania w razie potrzeby i Cialis do stosowania raz na dobę.
Cialis nie jest przeznaczony dla kobiet ani dzieci. Ważne jest, aby pamiętać, że leku Cialis nie należy przyjmować z lekami zwanymi “azotanami”, takimi jak dinitrat izosorbidu lub monoazotan izosorbidu, które są często przepisywane na ból w klatce piersiowej; lub z lekami rekreacyjnymi zwanymi “poppers”, takimi jak azotyn amylu lub butylu, ponieważ takie połączenie może spowodować niebezpieczny spadek ciśnienia krwi; lub w przypadku uczulenia na lek Cialis lub Adcirca® (tadalafil), lub którykolwiek z ich składników. Każda osoba, u której wystąpią jakiekolwiek objawy reakcji alergicznej, takie jak wysypka, pokrzywka, obrzęk warg, języka lub gardła, trudności w oddychaniu lub połykaniu, powinna natychmiast skontaktować się z lekarzem lub uzyskać pomoc.
Tyle w temacie komunikatu prasowego z dnia 30.05.2022 dotyczącego jednego z najbardziej znanych leków na zaburzenia erekcji na świecie.
Inny znany lek to Viagra. Sildenafil (Viagra), przykład leku o zmienionym przeznaczeniu, został wprowadzony na rynek jako lek przeciw dusznicy bolesnej. Ale jego stosowanie jest wykorzystywane jako lek na zaburzenia erekcji.
W Polsce jeden z leków na zaburzenia erekcji można kupić bez recepty, dzięki pozytywnemu rozpatrzeniu wniosku o przekształcenie leku na receptę w lek bez recepty. Szczęściarzem, któremu ten zabieg się udał jest firma Adamed Pharma.
Ale czy to wszystkie leki na zaburzenia erekcji?
Oczywiście, że nie!
Zaburzenia erekcji (Erectile dysfunction – ED) są najczęstszym problemem seksualnym u mężczyzn. ED definiuje się jako trudności w zainicjowaniu lub utrzymaniu erekcji prącia odpowiedniej do aktywności seksualnej. ED ma istotny wpływ na relacje intymne, jakość życia i ogólną samoocenę mężczyzn.
Ponadto, ED może być również wczesnym wskaźnikiem niewykrytej choroby sercowo-naczyniowej. Jedno z największych badań nad ED, Massachusetts Male Aging Study, wykazało, że częstość występowania ED wzrasta wraz z wiekiem i dotyczy nawet połowy populacji mężczyzn w wieku od 40 do 70 lat. Dlatego też, wraz ze wzrostem liczby osób starszych na świecie, szacuje się, że częstość występowania ED podwoi się z 152 milionów mężczyzn w 1995 roku do 322 milionów mężczyzn w 2025 roku. W większości udokumentowanych przypadków ED może również towarzyszyć nadciśnienie tętnicze, cukrzyca, otyłość i miażdżyca.
W latach 80. większość pionierskich badań nad ED zapoczątkowało wprowadzenie leków wazoaktywnych, które były bardzo skuteczne jako środki wywołujące sztywność prącia. Dopiero pod koniec lat 90. i na początku XXI wieku wprowadzono doustne inhibitory fosfodiesterazy-5, które odegrały istotną rolę w zrewolucjonizowaniu dziedziny medycyny seksualnej. Terapia pierwszego rzutu w leczeniu ED składa się z leków podawanych zarówno doustnie, jak i podjęzykowo, podczas gdy terapia polegająca na bezpośrednim wstrzykiwaniu środków wazoaktywnych do ciał jamistych (intravernosal injection, ICI) jest uważana jedynie za terapię drugiego rzutu.
Kiedy mężczyzna jest pobudzony seksualnie, mózg wysyła sygnał do zakończeń nerwowych w penisie, aby uwolnić tlenek azotu, który następnie działa za pośrednictwem enzymu, powodując wytworzenie substancji chemicznej zwanej cyklicznym monofosforanem guanozyny lub cGMP. To właśnie ta substancja chemiczna rozluźnia naczynia krwionośne w penisie, umożliwiając wystąpienie erekcji. Jednak w tym samym czasie inny związek, zwany fosfodiesterazą, powoduje, że cGMP staje się nieaktywny. U zdrowego mężczyzny pierwszy proces przeważa nad drugim i może dojść do erekcji.
Problemem mężczyzny z zaburzeniami erekcji jest to, że nie jest on w stanie wyprodukować wystarczającej ilości tlenku azotu, aby pierwszy proces mógł zdominować drugi.
Objaśnienie: cGMP (Cykliczny guanozyno-3′,5′-monofosforan) to cykliczny nukleotyd pochodzący z trifosforanu guanozyny (GTP). cGMP działa jak drugi przekaźnik, podobnie jak cykliczny AMP (cAMP-pochodna adenozynotrójfosforanu). Jego najbardziej prawdopodobnym mechanizmem działania jest aktywacja wewnątrzkomórkowych kinaz białkowych w odpowiedzi na wiązanie nieprzepuszczalnych przez błonę hormonów peptydowych z zewnętrzną powierzchnią komórki.
cGMP jest powszechnym regulatorem przewodnictwa kanałów jonowych, glikogenolizy i apoptozy komórek. Rozluźnia również tkankę mięśni gładkich. W naczyniach krwionośnych rozluźnienie mięśni gładkich naczyń prowadzi do rozszerzenia naczyń i zwiększenia przepływu krwi.
Anatomia prącia
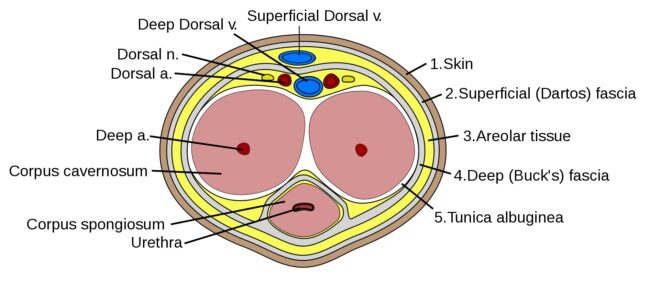
Prącie składa się z trzech równolegle biegnących ciał tkanki erekcyjnej: ciała gąbczastego (corpus spongiosum), obejmującego cewkę moczową i zakończonego żołędzią prącia, oraz dwóch ciał jamistych (corpora cavernosa – CC), które pełnią funkcję wypełnionych krwią kondensatorów zapewniających strukturę wzniesionego narządu. CC prącia są wysoce wyspecjalizowanymi strukturami naczyniowymi, które są morfologicznie przystosowane do pełnienia swojej funkcji, jaką jest nabrzmiewanie podczas podniecenia seksualnego. Mięsień gładki stanowi około 40-50% powierzchni przekroju poprzecznego tkanki, zgodnie z wynikami analizy histomorfometrycznej. W penisie znajdują się trzy główne tętnice: jamista, grzbietowa i opuszkowo-goleniowa. Wszystkie trzy wywodzą się ze wspólnego odgałęzienia tętnicy pudendalnej wewnętrznej i tworzą rozległą sieć anastomoz. Istnieje tendencja do przeprowadzania eksperymentów in vitro z użyciem tętnicy pudendalnej zamiast tętnicy jamistej w celu zbadania patofizjologicznych aspektów ED, ponieważ tętnica ta stanowi główny opór dla rozszerzenia prącia podczas stymulacji seksualnej. Nowe odkrycia sugerują, że tętnica pudendalna odpowiada za 70% całkowitego oporu naczyniowego prącia. Tętnice zaopatrujące CC w krew pochodzą głównie z tętnicy jamistej głębokiej prącia 9g, która powoduje, że ciało powiększenie podczas erekcji, podczas gdy tętnica grzbietowa głęboka powoduje powiększenie żołędzi. Drenaż żylny nie jest podobny do zaopatrzenia tętniczego; istnieje tylko jedna żyła grzbietowa głęboka, która biegnie obok tętnic grzbietowych i nerwów w powięzi Bucka powyżej błony białawej (tunica albuginea), która jest strukturą wielowarstwową, przez którą przechodzą żyły wysyłające. Układ żylny prącia u człowieka jest ogólnie opisywany jako pojedyncza żyła grzbietowa głęboka, której towarzyszy para tętnic grzbietowych, umiejscowionych pomiędzy powięzią ścięgnistą a powięzią Bucka w celu zapewnienia odpływu żylnego. Ciało gąbczaste (corpus spongiosum) jest tkanką erekcyjną analogiczną do CC, ale z cieńszą tunica albuginea (błona biaława – włóknista otoczka, która wydłuża długość ciał jamistych penisa). Cewka moczowa leży w obrębie ciała gąbczastego. Unerwienie prącia jest zarówno autonomiczne (współczulne i przywspółczulne), jak i somatyczne (czuciowe i ruchowe). Z neuronów w rdzeniu kręgowym i zwojach obwodowych nerwy współczulne i przywspółczulne łączą się, tworząc nerwy jamiste, które wnikają do CC i ciała gąbczastego, wpływając na procesy nerwowo-naczyniowe podczas tumescencji i detumescencji.
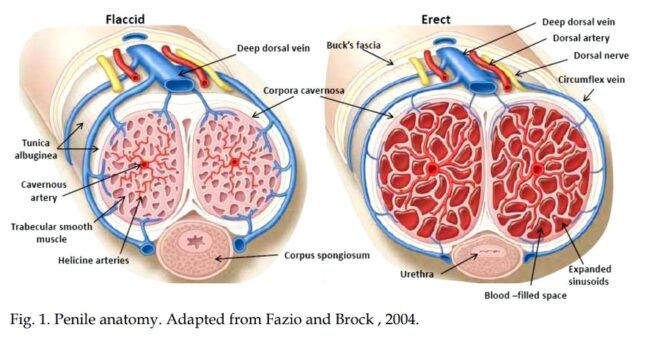
Fizjologia wzwodu prącia
Erekcja prącia (PE) obejmuje drogi centralne i obwodowe. Tumescencja jest inicjowana w wyniku centralnego przetwarzania i integracji bodźców dotykowych, wzrokowych, zapachowych i wyobrażeniowych. W wyniku stymulacji seksualnej generowane są sygnały do zaangażowanych tkanek obwodowych. Ostateczna reakcja jest zatem wynikiem skoordynowanej aktywności rdzenia kręgowego w drogach autonomicznych do prącia, a także w drogach somatycznych do mięśni prążkowanych krocza. Zarówno w ośrodkowej, jak i obwodowej regulacji PE bierze udział kilka neuroprzekaźników i układów, których szczegóły wciąż nie są do końca poznane. Wydaje się, że na poziomierdzeniowym istnieje sieć składająca się z pierwotnych aferentów z narządów płciowych, interneuronów rdzeniowych, jąder współczulnych, przywspółczulnych i somatycznych, która jest w stanie zintegrować wszystkie informacje. Obwodowo, równowaga między substancjami kontrolującymi stopień skurczu mięśni gładkich ciał jamistych.
Dynamiczne współdziałanie czynników zwężających i rozszerzających naczynia krwionośne w penisie decyduje o stanie wzwodu lub zwiotczenia.
PE jest uwarunkowany zmianami ciśnienia w tętniczkach i zatokach ciał jamistych. Naczynia mechanizmu erekcyjnego różnią się od większości łożysk naczyniowych, ponieważ składają się z tętniczek i zatok wypełnionych krwią, które są wyścielone mięśniami gładkimi i komórkami śródbłonka, jak opisano wcześniej. W stanie zwiotczenia tkanka ta jest tonicznie skurczona, co umożliwia jedynie niewielki przepływ tętniczy dla celów odżywczych. Ciśnienie parcjalne tlenu (PO2) we krwi wynosi około 35 mmHg. Z drugiej strony, rozszerzenie tętnic prącia jest pierwszym wydarzeniem w rozwoju erekcji. Jego konsekwencją jest wzrost przepływu krwi i ciśnienia w przestrzeni lacunarnej. Rozszerzenie zatok blokuje napływającą krew. Również odpływ żylny jest ograniczony przez ucisk przestrzeni żylnych między błoną białawą a zatokami obwodowymi. Powoduje to rozciągnięcie tuniki do granic jej możliwości i zmniejszenie odpływu żylnego do minimum, co prowadzi do wzrostu ciśnienia wewnątrzjamistego, które utrzymuje się na poziomie około l00 mmHg. Wzwód obejmuje zatem relaksację zatok, rozszerzenie tętnic i ucisk żył.
Przyczyny zaburzeń erekcji mogą być fizjologiczne lub psychologiczne.
Przyczyny zaburzeń erekcji:
Starzenie się
Uważa się, że starzenie się jest jedną z głównych przyczyn obniżonych funkcji seksualnych, na które wpływa również zmiana stylu życia, zwiększony stres, depresja, cukrzyca i/lub inne zaburzenia metaboliczne i endokrynologiczne. Różne leki, takie jak antydepresanty, środki uspokajające, nasenne, antyandrogeny i leki przeciwnadciśnieniowe również mogą prowadzić do upośledzenia funkcji seksualnych. Istnieje ścisły związek między starzeniem się a zaburzeniami erekcji.
Stres oksydacyjny
Stres oksydacyjny (OS) jest jednym z głównych czynników przyczyniających się do zaburzeń erekcji.
Wpływ chorób układu krążenia, otyłości, zespołu metabolicznego na zaburzenia erekcji
Otyłość jest zwykle związana z ogólnie przyjętymi czynnikami ryzyka zaburzeń erekcji, takimi jak nadciśnienie, hiperlipidemia i cukrzyca, ale ostatnio została sklasyfikowana jako niezależna przyczyna zaburzeń erekcji.
Palenie
Przeprowadzono kilka badań w celu potwierdzenia, że palenie jest niezależnym czynnikiem ryzyka zaburzeń erekcji.
Cukrzyca
Przez lata cukrzyca była znana jako jedna z głównych bezpośrednich przyczyn zaburzeń erekcji. Badania wykazały, że prawdopodobieństwo wystąpienia zaburzeń erekcji jest wyższe u mężczyzn z cukrzycą niż u mężczyzn bez cukrzycy w tym samym wieku i różnica ta rośnie wraz z wiekiem.
Wpływ regulacji hormonalnej na zaburzenia erekcji
Może istnieć związek między insulinoopornością, dysfunkcją śródbłonka, zespołem metabolicznym, ED i cukrzycą. Wykazano, że hipogonadyzm jest niezależną determinantą dysfunkcji śródbłonka, przyczyniając się w ten sposób do patologii naczyń, w tym ED. Testosteron (T) i jego metabolity, dihydrotestosteron (DHT) i estradiol (E2), odgrywają kluczową rolę w rozwoju i utrzymaniu prawidłowych męskich narządów płciowych, jąder, dodatkowych narządów płciowych, masy mięśni szkieletowych, masy wzrostu kości, męskiego owłosienia, libido i funkcji erekcyjnych. Uważa się również, że testosteron wpływa na identyfikację płci w ośrodkowym układzie nerwowym. Zarówno DHT, jak i testosteron mogą utrzymywać libido i erekcję, co wskazuje, że estrogeny nie są wymagane do ich utrzymania u mężczyzn. Receptory androgenowe (androgen receptors, ARs) są obecne w amygdali, przegrodzie bocznej i ciałach przedwzgórzowych u samców naczelnych. Wydaje się, że miejsca w mózgu związane z AR w podwzgórzu, przysadce mózgowej i obszarach preoptycznych mogą wpływać na męskie zachowania seksualne. Na przykład stymulacja jąder przodomózgowia, hipokampa i podwzgórza powoduje erekcję prącia i/lub zachowania godowe u zwierząt laboratoryjnych. Inne badania wskazują, że jądra przyzwojowe podwzgórza mogą być głównym źródłem zstępującej rdzeniowej drogi erekcji do rdzeniowego generatora erekcji.
Zmniejszona produkcja testosteronu może zwiększać ryzyko osteoporozy, dysfunkcji seksualnych, zmęczenia, chorób układu krążenia i zaburzeń nastroju oraz może zmniejszać masę mięśniową.
Postępowanie i leczenie zaburzeń erekcji
Zaburzenia erekcji to wada prącia polegająca na braku możliwości osiągnięcia i utrzymania erekcji z powodu czynników fizjologicznych lub psychologicznych. Zaproponowano różne metody leczenia, w tym:
- Terapia psychologiczna/behawioralna z udziałem przeszkolonego doradcy, której celem jest pomoc w radzeniu sobie z uczuciami niepokoju, strachu i poczucia winy, które mogą mieć wpływ na funkcje seksualne;
- Leczenie farmakologiczne i farmakologiczne (np. terapia zastępcza testosteronem w przypadku niewydolności androgenowej);
- Leczenie fitomedyczne;
- Leczenie chirurgiczne.
Leczenie pierwszego rzutu: Farmakoterapia doustna
Inhibitory PDE5
Rodzina fosfodiesteraz składa się z 11 enzymów katalitycznych, które regulują aktywność drugiego komunikatora w komórkach poprzez rozszczepienie wiązania fosfodiestrowego cyklicznego adenozynomonofosforanu (cAMP) lub cyklicznego monofosforanu guanozyny (cGMP), lub obu tych związków PDE5 występuje w wysokich stężeniach w mięśniach gładkich ciał jamistych prącia oraz w mięśniach gładkich tętnic krezkowych.
Inhibitory PDE5 są podstawową metodą farmakoterapii zaburzeń erekcji (ED).
PDE są środkami aktywnymi doustnie, które przyjmuje się na żądanie przed stosunkiem płciowym.
PDE5 został odkryty przez Corbina i współpracowników. Enzym PDE5 jest homo-dimerem z dwoma identycznymi podjednostkami, a każda podjednostka ma masę 100 000 kilodaltonów. Obie podjednostki mają domenę katalityczną i domenę regulatorową; jednak to domena katalityczna jest głównym celem inhibitorów PDE5. Niemniej jednak to właśnie odkrycie tlenku azotu (NO) i cGMP jako głównych efektorów w relaksacji mięśni gładkich prącia doprowadziło do identyfikacji inhibitorów PDE5, które mogą dodatkowo podnosić wewnątrzkomórkowe stężenie cGMP.
Większość inhibitorów PDE5 ma strukturę porównywalną do syldenafilu; ponadto część struktury molekularnej większości inhibitorów PDE5 przypomina strukturę cGMP. To ma znaczenie, ponieważ inhibitory PDE5 są konkurencyjnymi antagonistami zarówno cGMP, jak i PDE5.
Działania inhibitorów PDE5 są często opisywane w kategoriach ich selektywności, gdy porównuje się je z innymi inhibitorami PDE.
Inhibitory PDE5 są degradowane w wątrobie, ponieważ nie są metabolizowane przez żadne inne enzymy w komórkach mięśni gładkich ciał jamistych. Tak więc inhibitory PDE5 muszą być transportowane do wątroby, aby mogły zostać zdegradowane. Należy zwrócić na to uwagę, ponieważ decyduje to o czasie działania, ponieważ zniknięcie inhibitora z osocza może wskazywać, ale nie zawsze dowodzi, na jego usunięcie z komórek, w których wywołał efekty.
Osoby otrzymujące inhibitory PDE5 zgłaszają erekcje wystarczające do odbycia stosunku płciowego . Inhibitory PDE5 wydają się jednak być mniej skuteczne w badaniach, w których u mężczyzn występują choroby współistniejące, takie jak cukrzyca.
Mimo to inhibitory PDE5 pozostają podstawową linią leczenia doustnego, a do najczęściej zgłaszanych działań niepożądanych należą: bóle głowy, dyspepsja i niedrożność nosa. Jeśli chodzi o bezpieczeństwo sercowo-naczyniowe (CV), inhibitory PDE5 okazały się bezpieczne u pacjentów z chorobą CV; jednak ze względu na mechanizm działania inhibitory PDE5 są przeciwwskazane u pacjentów z ED przyjmujących azotany z powodu nieprzewidywalnego niedociśnienia.
Syldenafil
Od czasu wprowadzenia sildenafilu jako skutecznej podstawy leczenia ED w 1998 roku, podobne środki zostały poddane badaniom klinicznym, a następnie wprowadzone do praktyki klinicznej. Istnieją niezbite dowody na to, że syldenafil jest skuteczny w leczeniu ED w szerokiej populacji mężczyzn. W rezultacie sildenafil okazał się skuteczny w przypadkach, gdy ED jest konsekwencją chorób współistniejących, takich jak cukrzyca, depresja, choroby układu sercowo-naczyniowego, nadciśnienie tętnicze i zakażenia dolnych dróg moczowych.
Sildenafil wykazuje skuteczność w dawkach 25, 50 lub 100 mg, a początek działania następuje zwykle w ciągu 25-60 minut; jednakże wchłanianie leku jest spowolnione w przypadku jedzenia. Podczas stosowania syldenafilu występują niewielkie działania niepożądane; najczęściej zgłaszanymi działaniami niepożądanymi były bóle głowy, zaczerwienienie, niestrawność i zmiany widzenia.
Udenafil (DA-8159)
Udenafil (DA-8159) jest inhibitorem PDE5; jest lekiem długo działającym o czasie półtrwania (t1/2) wynoszącym 11- 13 godzin i charakteryzuje się stosunkowo szybkim wchłanianiem – stężenie w osoczu po spożyciu osiąga tmax, czyli szczytowe stężenie leku, w ciągu 1-1,5 godziny. Dane z badania klinicznego II fazy wykazały, że u mężczyzn z łagodną do ciężkiej postacią ED udenafil spowodował znaczną poprawę funkcji erekcji po 12 tygodniach leczenia. W badaniu III fazy przeprowadzonym w Korei oceniano skuteczność i bezpieczeństwo stosowania udenafilu u pacjentów z ED. Wszystkie odpowiedzi na udenafil były istotnie większe niż w przypadku placebo (p < 0,0001), a pacjenci otrzymujący udenafil w dawkach 100 mg lub 200 mg byli istotnie (p < 0,0001) bardziej zadowoleni ze swojego życia seksualnego w porównaniu z mężczyznami przyjmującymi placebo. W badaniach na zwierzętach podawanie DA-8159 (0,3 lub 1 mg/kg) wywoływało zależny od dawki wzrost ciśnienia wewnątrzjamowego (ICP); przewlekłe leczenie DA-8159 przywracało reakcje erekcyjne wywołane elektryczną stymulacją, poprawiało funkcję śródbłonka i znacząco zmniejszało stężenie w osoczu zarówno endoteliny (silnego czynnika zwężającego naczynia), jak i asymetrycznej dimetyloargininy (ADMA) (inhibitora wytwarzania NO). Udenafil był dobrze tolerowany, ponieważ większość działań niepożądanych stanowiły: zaczerwienienie, rozstrój żołądka i bóle głowy, które były na ogół łagodne lub umiarkowane.
Awanafil
Awanafil jest pochodną pirymidyny zsyntetyzowaną jako wysoce selektywny inhibitor PDE5 do leczenia ED. Badania wykazały, że do 84% dawek awanafilu powodowało wystarczającą erekcję do aktywności seksualnej, w porównaniu do placebo. W badaniach klinicznych awanafil wykazał większą selektywność, prawie 120-krotnie, wobec PDE6 niż syldenafil i wardenafil.
Dodatkowo, w porównaniu do syldenafilu, wardenafilu i tadalafilu (opisanych później), struktura chemiczna awanafilu różni się od standardowego modelu diestru nukleinowego baza/cukry/fosforan; struktura molekularna awanafilu jest pochodną azotu karboksyamidu pirymidynowego, w którym atom azotu podstawnika amidowego jest związany z grupą pirymidynylometylowa. W wyniku swojej unikalnej struktury chemicznej, teoretycznie, awanafil może wiązać się z miejscem katalitycznym PDE5 niezależnie od orientacji przestrzennej podstawnika amidowego.
W ocenie farmakokinetycznej tego leku w badaniach odnotowano szybkie wchłanianie z tmax około 35 minut i krótkim t1/2 poniżej 1,5 godziny, bez niepożądanej kumulacji leku. Korzystne dane z badania interakcji azotanów wykazały jedynie niewielki wpływ na ciśnienie krwi i częstość akcji serca.
Lodenafil
Węglan lodenafilu jest nowszym inhibitorem PDE5; jest to dimer utworzony przez dwie cząsteczki lodenafilu połączone mostkiem węglanowym. Po połknięciu, mostek węglanowy zostaje przerwany dostarczając aktywny związek lodenafil. Działanie lodenafilu było szeroko badane in vitro; zauważono, że lek ten powoduje zależne od stężenia rozluźnienie tkanki ciał jamistych prącia królika i człowieka poprzez wzmocnienie zależnego od NO- rozluźnienia tkanki prącia w odpowiedzi na acetylocholinę lub transmuralną stymulację polem elektrycznym (EFS). Dla porównania wykazano, że lodenafil jest około dwukrotnie silniejszy od syldenafilu w hamowaniu rozkładu cGMP. Zbadano również jego skuteczność i bezpieczeństwo stosowania w dawkach 20 mg, 40 mg i 80 mg i wykazano, że znacząco poprawia wyniki domeny erekcji Międzynarodowego Indeksu Funkcji Erekcyjnych (IIEF domain scores: szeroko stosowany, wielowymiarowy instrument samoopisowy do oceny funkcji seksualnych mężczyzn), przy jedynie łagodnych lub umiarkowanych działaniach niepożądanych, takich jak ból głowy, zaczerwienienie, zaburzenia widzenia i dyspepsja. Lodenafil jest atrakcyjnym środkiem farmakoterapii w leczeniu ED.
Tadalafil
Tadalafil okazał się skuteczny w wielu specjalnych populacjach mężczyzn, u których ED jest spowodowana różnymi schorzeniami, w tym cukrzycą, radioterapią z powodu raka prostaty, urazem rdzenia kręgowego i zakażeniem dolnych dróg moczowych.
Tadalafil stosowany w dawkach 10 i 20 mg znacząco poprawiał erekcję i był dobrze tolerowany, ponieważ jedynymi zgłaszanymi działaniami niepożądanymi były: ból głowy, zaczerwienienie, niestrawność, przekrwienie nosa i ból pleców lub obręczy. Nie ma przekonujących dowodów na jakiekolwiek istotne kwestie bezpieczeństwa dotyczące przeciwwskazań sercowo-naczyniowych przy stosowaniu tadalafilu w zalecanych dawkach 2,5 mg i 5 mg na dobę.
Wardenafil
Vardenafil jest skutecznym środkiem wazoaktywnym w leczeniu ED w szerokiej populacji w dawkach 10 mg i 20 mg. Vardenafil był nawet w stanie wywołać odpowiednią erekcję do aktywności seksualnej w specjalnych populacjach, gdzie ED były wynikiem cukrzycy, chemioterapii, depresji, nadciśnienia i urazu rdzenia kręgowego. Mechanizm działania wardenafilu jest podobny do innych inhibitorów PDE5, dlatego jego działania niepożądane są podobne do tych zgłaszanych przez inne inhibitory PDE5, takich jak ból głowy, zaczerwienienie, niestrawność i zatkanie nosa.
Ogólnie rzecz biorąc, wardenafil pozostaje wysoce zalecanym środkiem wazoaktywnym, ponieważ udowodniono jego korzystne działanie u wielu pacjentów z ED, a nie ma przekonujących dowodów na jakiekolwiek istotne kwestie bezpieczeństwa, dotyczące przeciwwskazań sercowo-naczyniowych, związanych z jego stosowaniem.
Trazodon
Trazodon jest “atypowym” lekiem przeciwdepresyjnym i selektywnie hamuje ośrodkowy wychwyt 5- hydroksytryptaminy (5-HT) poprzez zwiększenie obrotu dopaminami w mózgu.
Trazodon ma działanie blokujące receptory alfa-adrenergiczne (α-AR) i, wraz ze swoim metabolitem meta- chlorofenylopiperazyną (m-CCP), trazodon jest znany z wywoływania erekcji w badaniach na zwierzętach poprzez selektywne blokowanie receptorów α-AR i zwiększanie spontanicznej częstotliwości odpalania nerwów w ciałach jamistych. Pomimo obiecującego sposobu działania w obrębie tkanki ciał jamistych, trazodon nie jest skutecznym lekiem u większości mężczyzn z ED, ponieważ doustne podawanie trazodonu wiąże się z dużą częstością priapizmu. Co więcej, w podwójnie ślepym badaniu z placebo trazodon nie był skuteczny nawet w dużych dawkach 150 do 200 mg na dobę.
Trazodon jest jednak nadal realną opcją dla mężczyzn z psychogenną ED, wynikającą z lęku lub depresji.
Agoniści receptora melanokortyny (PT-141)
PT-141 był początkowo produkowany jako środek do opalania; odkryto jednak, że po wstrzyknięciu podskórnym inicjuje silną erekcję u mężczyzn z nieorganiczną ED. PT-141 jest syntetycznym, cyklicznym, nieselektywnym agonistą receptorów melanokortyny, o wysokim powinowactwie do receptorów MC 1, 3 i 4, i uważa się, że jest metabolitem melanotanu-II (MT-II).
Teoretycznie agoniści receptorów MC mogą mieć korzystne działanie u pacjentów z ED na podstawie wyników podwójnie ślepego, kontrolowanego placebo badania, w którym PT-141, w dawkach od 4 do 20 mg, podawano 32 zdrowym osobom.
Wyniki tego badania wykazały, że PT-141 znacząco zwiększał aktywność erekcji nawet bez wizualnej stymulacji seksualnej, w porównaniu z osobami otrzymującymi placebo. Ponadto, w kontrolowanym placebo badaniu crossover, u mężczyzn z łagodną do umiarkowanej ED, którym podawano PT-141 wraz z wizualną stymulacją seksualną, odnotowano trzykrotny wzrost aktywności erekcyjnej w porównaniu z placebo. Jednoczesne stosowanie PT-141 z syldenafilem w dawce 25 mg znacząco zwiększyło sztywność prącia w porównaniu z samym syldenafilem. Zauważono również, że gdy pacjenci przyjmowali oba leki w skojarzeniu, nie obserwowano znaczącego wzrostu działań niepożądanych w porównaniu z tymi, które występowały podczas przyjmowania samego syldenafilu lub PT-141.
Leki otwierające kanały potasowe
Leki otwierające kanały potasowe, takie jak pinacidil, kromakalim, lemakalim i nikorandil, okazały się skuteczne w osiąganiu erekcji u mężczyzn z ED. Otwieracze kanału potasowego działają poprzez hiperpolaryzację błony komórkowej, co zwiększa przepuszczalność błony komórkowej dla jonów potasu, powodując jej rozluźnienie i w konsekwencji erekcję. Doświadczenie kliniczne z tymi lekami otwierającymi kanały potasowe w leczeniu ED jest ograniczone; dlatego leki będące agonistami kanałów potasowych nie zostały zatwierdzone w kontrolowanych badaniach klinicznych jako alternatywna metoda leczenia mężczyzn z ED.
Inhibitory kinazy Rho
Skurcz i rozkurcz mięśni gładkich jest związany z poziomem wolnego wapnia cytozolowego, który jest częściowo regulowany przez akumulację wewnątrzkomórkowych wtórnych posłańców, takich jak trifosforan inozytolu (IP3 ) i diacyloglicerol (DAG) poprzez aktywację fosfolipazy C (PLC)
. Aktywacja PLC ułatwia uwalnianie i wzrost stężenia wewnątrzkomórkowego Ca2+ co powoduje wiązanie wapnia z kalmoduliną i następczą aktywację kinazy łańcucha lekkiego miozyny. Kiedy wewnątrzkomórkowy poziom wapnia obniża się, RhoA, małe monomeryczne białko G, aktywuje Rho-kinazę; z kolei Rho-kinaza fosforyluje i jednocześnie hamuje regulatorową podjednostkę fosfatazy lekkiego łańcucha miozyny.
Tak więc Rho-kinazę uważa się za niezbędną w systemie uwrażliwiania komórek na wapń, ponieważ jej aktywacja tworzy kaskadę, w której fosforylacja łańcuchów lekkich miozyny wyzwala cykle mostków poprzecznych miozyny wzdłuż filamentów aktynowych, generując siłę skurczu, która jest utrzymywana, o ile nie zostanie zahamowana, w obrębie ciał jamistych. Chitaley, K. i wsp. po raz pierwszy wykazali, że Rho-kinaza przyczynia się do napięcia mięśni gładkich w ciałach jamistych. Wstrzyknięcie inhibitora Rho-kinazy Y-27632 do zatok ciał jamistych spowodowało wzrost ciśnienia wewnątrzjamistego w ciągu kilku minut w sposób zależny od dawki, bez znaczącego obniżenia systemowego ciśnienia krwi. Co więcej, wzrost ciśnienia wewnątrzjamistego pod wpływem inhibicji Rho był niezależny od relaksującego działania NO i cyklazy guanylanowej, co wskazuje, że szlak kinazy Rho może być alternatywnym celem terapeutycznym w leczeniu ED. W badaniach na zwierzętach inhibitor Rho-kinazy, fasudil, był skuteczny w zwalczaniu ED o podłożu naczyniowym, a ponadto był w stanie zmniejszyć poziom miażdżycy miednicy. W badaniu dotyczącym ED związanych z cukrzycą u szczurów wykazano, że przewlekłe podawanie fasudilu było skuteczniejsze niż inne popularne metody leczenia ED, w odwracaniu szkodliwych zmian biochemicznych wywołanych wysokim poziomem insuliny.
Badania kliniczne na ludziach wykazały, że atorwastatyna (Lipitor) w małej dawce (lek działający poprzez hamowanie reduktazy HMG-CoA, enzymu występującego w tkance wątrobowej, który odgrywa kluczową rolę w produkcji cholesterolu w organizmie) normalizuje odpowiedź cukrzycy na syldenafil u szczurów z cukrzycą leczonych streptozotocyną (STZ). Wynik ten sugeruje, że statyny mogą mieć również działanie hamujące w mechanizmie RhoA/Rho-kinazy.
Alternatywne metody leczenia ED
ZIOŁOWE LECZENIE ED (johimbina)
Chociaż leki ziołowe nie są przepisywane z wyboru pacjentom w innych częściach świata, takich jak Azja, Afryka i Bliski Wschód, są one szeroko stosowaną formą terapii dla mężczyzn z ED. Jednakże, mimo że istnieje mnóstwo ziołowych metod leczenia, które są bardzo atrakcyjne dla pacjentów z ED, należy zachować ostrożność, ponieważ niektóre leki ziołowe mogą powodować szkodliwe interakcje lekowe, zwłaszcza u mężczyzn z chorobami współistniejącymi, takimi jak choroby układu krążenia i cukrzyca. Z drugiej strony, ziołowe lub tradycyjne podejście terapeutyczne może być bardziej akceptowane kulturowo przez niektórych pacjentów, którzy mogą preferować tę holistyczną opcję ze względu na preferencje religijne, społeczne, finansowe lub inne osobiste.
W rezultacie rośnie zainteresowanie społeczeństwa alternatywnymi, holistycznymi metodami leczenia, co spowodowało wzrost zapotrzebowania na takie terapie.
Chociaż dostępnych jest wiele leków ziołowych stosowanych w ED, w niniejszym przeglądzie skoncentrujemy się na skuteczności johimbiny.
Yohimbina jest alkaloidem pozyskiwanym z afrykańskiego drzewa yohimbe (Pausinystalia yohimbe).
Yohimbina została dobrze scharakteryzowana jako antagonista receptora adrenergicznego alfa-2 (α2 -AR). Specyficznie hamuje ona presynaptyczne receptory α-2 adrenergiczne w mózgu; w rezultacie dochodzi do zmniejszenia napięcia współczulnego, ponieważ stężenie noradrenaliny w mózgu i rdzeniu kręgowym ulega zmniejszeniu. Stwierdzono, że okres półtrwania johimbiny w osoczu wynosi 0,6 godziny, natomiast noradrenergiczne działanie leku w osoczu utrzymywało się przez ponad 12 godzin.
Teoretycznie johimbina powinna być bardzo skutecznym lekiem w leczeniu ED, jednak w kilku kontrolowanych badaniach z udziałem pacjentów z różnymi rodzajami ED johimbina przyniosła jedynie umiarkowane efekty. Jednak kompleksowy przegląd systematyczny wykazał przewagę johimbiny nad placebo w leczeniu ED.
W połączeniu z innymi lekami, doustna johimbina (15 mg dziennie) z trazodonem (50 mg dziennie) okazała się bezpiecznym i skutecznym leczeniem psychogennych ED. Pomimo rosnącej popularności, johimbina nie jest zalecana w większości wytycznych dotyczących leczenia ED, ze względu na sprzeczne doniesienia kliniczne dotyczące jej skuteczności.
Terapia antyoksydacyjna (kwercetyna)
Stres oksydacyjny występuje w przypadku braku równowagi między prooksydantami a zdolnością antyoksydantów do wymiatania reaktywnych form tlenu (ROS). ROS szybko inaktywują NO, tym samym ograniczając jego zdolność do relaksacji mięśni gładkich. Badania wykazały również, że ROS odgrywają główną rolę w indukowaniu apoptozy w tkankach ciał jamistych. Tak więc, istnieją przytłaczające dowody wskazujące na rolę stresu oksydacyjnego w patofizjologii ED. W związku z tym leczenie antyoksydacyjne, mające na celu regulację ROS, jest badane jako potencjalna terapia ED.
Kwercetyna (pentahydroksyflawon) jest najliczniej występującym flawonoidem w diecie człowieka i jest dostępna bez recepty jako suplement diety. Flawonoidy to roślinne związki fenolowe o silnych właściwościach przeciwutleniających, występujące w różnych źródłach pokarmowych, takich jak herbata, cebula i brokuły. Kwercetyna jest silnym przeciwutleniaczem, a jej mechanizm działania polega na bezpośrednim wymiataniu wolnych rodników, takich jak nadtlenek wodoru i nadtlenek, hamowaniu prooksydacyjnego enzymu oksydazy ksantynowej, hamowaniu peroksydacji lipidów, chelatowaniu żelaza oraz zmianie szlaków obrony antyoksydacyjnej w komórce.
Biorąc pod uwagę jej korzystne działanie przeciwko uszkodzeniom oksydacyjnym, uznano, że kwercetyna może być skuteczna w leczeniu ED, w szczególności ED w wyniku cukrzycy. Badania na zwierzętach wykazały, że leczenie kwercetyną skutecznie poprawiło ciśnienie wewnątrzjamiste szczurów z cukrzycą poprzez zachowanie aktywności dysmutazy ponadtlenkowej (SOD), (enzymu antyoksydacyjnego), przy jednoczesnym zwiększeniu ekspresji śródbłonkowej syntazy tlenku azotu (eNOS). Wyniki wyraźnie pokazują, że ekspresja eNOS i poziomy NO w ciałach jamistych były znacząco zwiększone u szczurów z cukrzycą, w odpowiedzi na leczenie kwercetyną.
Leczenie drugiego rzutu: Farmakoterapia wewnątrzjamowa
ICI (Intracavernosal injection) to terapia drugiego rzutu, którą rozważa się głównie u pacjentów, u których nie uzyskano odpowiedzi na terapię pierwszego rzutu lub u tych, którzy nie mogą stosować najmniej inwazyjnych form dostępnej farmakoterapii. Idealnymi kandydatami do terapii ICI są pacjenci, którzy używają azotanów lub mogliby potencjalnie używać azotanów, mają uszkodzenie nerwów w wyniku operacji miednicy, urazów, pacjenci z cukrzycą oraz pacjenci, którzy pragną szybkiego początku erekcji, większej sztywności i czasu trwania erekcji. Przeciwwskazaniami do terapii ICI są pacjenci z wywiadem w kierunku priapizmu po stosowaniu leków wazoaktywnych oraz pacjenci z ciężkim zwłóknieniem prącia po stosowaniu inhibitorów monoaminooksydazy (MAOI); ponadto brak odpowiedzi na ICI występuje najczęściej w wyniku zastosowania nieodpowiedniej dawki, błędnego wstrzyknięcia podskórnego lub dożylnego.
Po ICI i stymulacji seksualnej sztywność prącia osiągana jest zwykle w ciągu 5-10 minut, ponieważ szybki wzrost przepływu krwi w tętnicach jamistych prowadzi do ucisku podwpustowych żył znajdujących się pomiędzy mięśniem gładkim ciał jamistych a wewnętrzną tunica albuginea (penis). Przy zablokowanym odpływie żylnym ciśnienie wewnątrzjamiste może być większe niż 100 mmHg. Skurcz naczyń krwionośnych i utrata sztywności prącia są wynikiem wzrostu napięcia współczulnego w czasie ejakulacji. W terapii ICI najczęściej przepisywanymi środkami wazoaktywnymi są prostaglandyna E-1 (PGE-1), fentolamina i papaweryna. Jednak wraz z postępem badań medycznych dostępne są inne skuteczne metody leczenia, takie jak wazoaktywny polipeptyd jelitowy, peptyd związany z genem kalcytoniny i aktywatory cyklazy guanylowej.
Papaweryna
Papaweryna została odkryta przez Mercka w 1848 roku, jest alkaloidem opium pochodzącym z maku lekarskiego Papaver somniferum. W leczeniu ED, wewnątrzjamowe wstrzyknięcie papaweryny było pierwszym środkiem farmakologicznym, który okazał się być klinicznie skuteczną terapią ED.
Papaweryna wywołuje rozluźnienie tkanki mięśniowej gładkiej ciał jamistych i tętnicy ramiennej, co prowadzi do erekcji prącia poprzez nieswoiste hamowanie fosfodiesterazy, co prowadzi do zwiększenia wewnątrzkomórkowego poziomu cGMP i cyklicznego adenozynomonofosforanu (cAMP).
W badaniach in vitro wykazano, że papaweryna zwiększa relaksację nie tylko w izolowanych pasmach mięśni gładkich ciał jamistych, ale również w tętnicach prącia i żyłach prącia; ponadto badania te wykazały również, że papaweryna była w stanie osłabić skurcze wywołane przez stymulację nerwów adrenergicznych i egzogenną noradrenalinę. Papaweryna może również regulować napięcie mięśni gładkich ciał jamistych drogą niezależną od cAMP poprzez hamowanie zależnych od napięcia kanałów wapniowych typu L.
Chociaż papaweryna jest skutecznym i niedrogim lekiem w leczeniu ED, w zestawionych doniesieniach wykazano, że stosowanie papaweryny wiąże się ze zwiększoną częstością występowania priapizmu(bolesnego wzwodu członka) i zwłóknienia. Po podaniu papaweryny bardzo często występuje również ból prącia.
Papaweryna może powodować inne działania niepożądane, takie jak: zaczerwienienie skóry, pocenie się, rozstrój żołądka, utrata apetytu, biegunka, zaparcia i nieregularne bicie serca.
Antagonista receptora alfa-adrenergicznego (fentolamina)
Chociaż korzystne działanie wazoaktywne fentolaminy zostało odkryte w latach 70. XX wieku, obecne zastosowanie kliniczne tego leku ogranicza się do stosowania go jako składnika innych leków wazoaktywnych. W ciałach jamistych prącia występują co najmniej trzy ważne izoformy alfa-adrenoreceptorów: 1A, 1B i 1D. Białka 1A i 1D zostały zidentyfikowane jako dominujące podtypy w ciałach jamistych prącia. Jednak rozróżnienie i rola poszczególnych podtypów receptora w szlakach sygnalizacji komórkowej w funkcji erekcji pozostaje w większości przypadków nieokreślona.
Receptory alfa-1 i 2-adrenoreceptory są zaliczane do receptorów sprzężonych z białkami G, które inicjują kilka złożonych procesów. Sugeruje się, że receptory alfa 1 utrzymują napięcie skurczowe prącia poprzez zwiększenie poziomu wapnia wewnątrzkomórkowego, po którym następuje napływ wapnia zewnątrzkomórkowego. Należy również zauważyć, że system receptorów -adrenergicznych odgrywa kluczową rolę w modulacji wiotkości prącia. Skurcz indukowany przez postjonalne receptory 2-adrenergiczne jest spowodowany zwiększeniem stężenia wapnia wewnątrzkomórkowego, ale obejmuje również rekrutację kanałów jonowych, aktywację fosfodiesteraz i tłumienie aktywności cyklazy adenylanowej.
Mesylan fentolaminy wywołuje blokadę receptorów -adrenergicznych; jest nieselektywnym – antagonistą adrenoreceptorów o podobnym powinowactwie do 1 i 2-adrenoreceptorów; ma również bezpośrednie, dodatnie działanie inotropowe i chronotropowe na mięsień sercowy, a także działanie wazodylatacyjne na mięśnie gładkie naczyń. Mimo że ICI fentolaminy zwiększa cielesny przepływ krwi, jednoczesny wzrost stężenia noradrenaliny zapobiega rozkurczowi zatok.
Badania wykazały również, że fentolamina wywołuje relaksację ciał jamistych erekcyjnych niezależnie od blokady 1 i 2-adrenoreceptorów, za pośrednictwem mechanizmu noradrenergicznego, pośredniczonego przez śródbłonek, sugerującego aktywację syntazy NO.
Prostaglandyna E1
Prostaglandyny zostały po raz pierwszy scharakteryzowane przez Bergstroma, S., Bengta, S. i Vane’a, J., jednak dopiero w 1986 r. Ishii, N. i Adaikan, PG opisali wazoaktywne działanie prostaglandyny E1s (PGE-1) do stosowania wewnątrzjamistego. W 1996 r. PGE-1 stała się pierwszym zatwierdzonym przez FDA wazoaktywnym lekiem do stosowania wewnątrzjamistego w leczeniu ED, a jej skuteczność została potwierdzona w trzech wieloośrodkowych, randomizowanych, prospektywnych badaniach klinicznych z sześciomiesięcznym przedłużeniem badania w trybie otwartym. Od tego czasu zgromadzono obszerną wiedzę na temat skuteczności i mechanizmu działania tego leku. PGE-1 jest jednym z najbardziej popularnych środków wazoaktywnych w terapii ICI.
PGE-1 indukuje rozkurcz mięśni gładkich ciał jamistych prącia poprzez regulację aktywności cyklazy adenylanowej, zwiększenie produkcji cAMP w prąciu poprzez aktywację receptorów prostaglandynowych EP, a następnie doprowadzenie do zmniejszenia wewnątrzkomórkowego stężenia wapnia, co skutkuje rozkurczem mięśni gładkich ciał jamistych. Rozszerzenie naczyń pod wpływem PGE- 1 odbywa się przez połączenia szczelinowe w obrębie prącia; dodatkowo PGE-1 hamuje aktywność współczulną poprzez działanie na neurony presynaptyczne, blokując uwalnianie noradrenaliny. PGE-1 jest metabolizowana przez 15-hydroksydehydrogenazę, a jej okres półtrwania w osoczu krwi wynosi mniej niż 1 minutę, ponieważ enzymy wątroby, nerek i prącia przyczyniają się do przekształcenia PGE-1 do jej nieaktywnej postaci. Częstość występowania priapizmu, potencjalnie niszczącego działania niepożądanego, jest niska przy stosowaniu PGE-1, co czyni ją najczęściej stosowanym środkiem wazoaktywnym w iniekcjach.
Wazoaktywny polipeptyd jelitowy
Wazoaktywny peptyd jelitowy (VIP) jest naturalnie występującym neuroprzekaźnikiem i silnym środkiem zwiotczającym mięśnie gładkie. VIP, który został wyizolowany z jelita cienkiego, został również po raz pierwszy opisany jako specyficzny neuroprzekaźnik wywołujący erekcję prącia w 1986 roku. Podczas gdy nerwy VIPergiczne są najgęściej skoncentrowane w prąciu wokół tętnic krezkowych, wstępne badania wewnątrzjamowe z zastosowaniem VIP dały niezadowalające wyniki; jednakże, gdy VIP-y były stosowane w połączeniu z lekami takimi jak papaweryna i fentolamina, obserwowano odpowiednie erekcje prącia. W związku z tym, terapia VIP-ami w ED jest stosowana głównie w połączeniu z mesylanem fentolaminy.
W działaniu VIP pośredniczy specyficzny receptor sprzężony z białkiem G (GPCR), który jest związany z cyklazą adenylanową. VIP łączy się z syntazą tlenku azotu (NOS) w obrębie włókien nerwowych okołonaczyniowych i trabekularnych unerwiających prącie, regulując w ten sposób ukrwienie prącia i napięcie mięśni gładkich męskich narządów płciowych. Większość NO i VIP-ergicznych nerwów ma charakter cholinergiczny, ponieważ zawierają one pęcherzykowy transporter acetylocholiny.
Biorąc pod uwagę, że działanie VIP jest bezpośrednio związane z cyklazą adenylanową, VIP może zwiększać stężenie cAMP w tkankach ciał jamistych bez wpływu na stężenie cGMP. VIP ma powszechnie obserwowane działania niepożądane, takie jak zaczerwienienie twarzy i bóle głowy, które są częstymi zdarzeniami charakterystycznymi dla większości terapii wazoaktywnych.
Chlorowodorek linsidominy
Donory tlenku azotu, takie jak nitroprusydek sodu (SNP) i linsidomina (SIN-1) mogą być stosowane w leczeniu ED. Badania in vivo wykazały, że SNP i linsidomina (SIN-1) zwiększają produkcję cGMP, co z kolei zwiększa uwalnianie NO w sposób zależny od dawki.
Uwalnianie NO powoduje rozluźnienie mięśni gładkich naczyń krwionośnych, a także mięśni gładkich cavernosum. Mimo że SIN-1 ma krótki okres półtrwania, wynoszący zaledwie 1-2 min, istnieje możliwość wywołania ogólnoustrojowego niedociśnienia przy stosowaniu tego leku. SNP i SIN-1 wykazują pozytywne efekty hemodynamiczne promujące funkcje erekcyjne i wykazują długoterminową obietnicę jako wazoaktywne środki do stosowania wewnątrzjamistego w porównaniu z innymi formami leczenia wewnątrzjamistego.
Peptyd związany z genem kalcytoniny
Peptyd związany z genem kalcytoniny (CGRP) powoduje wzrost napływu krwi do tętnic prącia, rozkurcz mięśni gładkich ciał jamistych i okluzję odpływu z ciał jamistych. Wykazano również, że CGRP, jako silny środek rozszerzający naczynia, wywołuje zależny od dawki wzrost przepływu krwi w prąciu przy pomocy agonistów receptorów specyficznych dla CGRP. W leczeniu ED, CGRP jest zwykle podawany razem z PGE-1, a ta terapia łączona okazała się skuteczna w poprawie funkcji erekcji.
Aktywator cyklazy guanylowej
Wykazano, że rozpuszczalne aktywatory cyklazy guanylanowej (sGC) zwiększają ciśnienie wewnątrzjamiste w tkance ciał jamistych . BAY41-2272 jest aktywatorem sGC, który, jak wykazano na stronie, rozluźnia ludzkie ciało jamiste z lub bez obecności NO. Enzym sGC przekształca GTP w cGMP, który jest aktywowany, gdy NO jest uwalniany z nerwów autonomicznych erekcji, co powoduje rozluźnienie mięśni gładkich. W penisie cGMP jest przekaźnikiem sygnałów komórkowych, który rozluźnia mięśnie gładkie ciał jamistych, powodując odpowiedni przepływ krwi niezbędny do erekcji prącia.
Autor: Quek Kia Fatt, Jason E. Davis, Pedrosa Nunes, Inger Stallmann-Jorgensen, R. Clinton Webb,Materiał Prasowy Sanofi -FDA. rys.wikipedia.org





- Znakomity
- Bardzo Dobry
- Dobry
- Przeciętny
- Słaby
- Beznadziejny






Więcej
Czy ludzie agresywni powinni być izolowani i co się dzieje z ich mózgiem?
Czy Polsce grozi epidemia Ćpunów?
Ci ludzie są tak głupi a może tak mądrzy albo chorzy?