
Ostatnia aktualizacja 28 lipca 2022
Tak jak w przypadku wydłużenia czasu trwania i powiększenia obszaru jego świętowania w tytule na który sobie prowokacyjnie pozwoliliśmy przy okazji „Europejskiego Dnia Mózgu”(wg kalendarium medycznego link 18 marca), który powinien trwać cały rok, tak Światowy Dzień Zespołu Downa (21 marca) jest okazją, aby co roku przypominać o tej niszowej i niewielu interesującej nieuleczalnej chorobie, choć rzucającej się gapiom w oczy. Nauka jak na razie jest bezbronna na tę przypadłość, ale przez ostatnie pół wieku coraz więcej o niej wiemy. Zespół Downa u poczętego dziecka jest również powodem znacznej ilości aborcji, dokonywanych po jego wykryciu – wskażnik powyżej 70%.
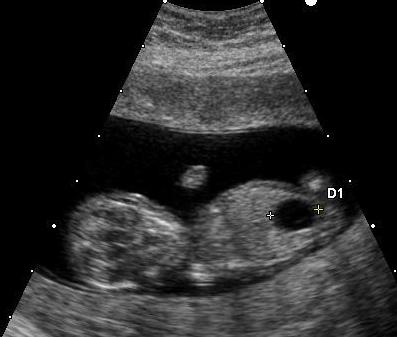
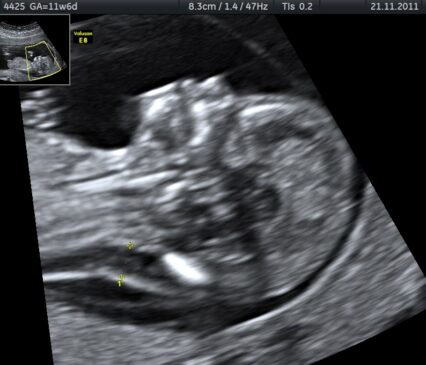
Nazwa pochodzi od Johna Langdona Downa, który był lekarzem brytyjskim, znanym z opisywania tej genetycznej choroby.
Zespół Downa jest jednym z najlepiej poznanych i najczęstszych zaburzeń chromosomowych spowodowanych obecnością trzeciej kopii chromosomu 21 (trisomia 21). Jest to najczęstsza genetyczna przyczyna upośledzenia umysłowego.
Częstość występowania zespołu Downa wynosi około 1/800 noworodków. Ryzyko urodzenia dziecka z zespołem Downa wzrasta wraz z wiekiem matki. Istnieje kilka cech, które występują w całej populacji osób z zespołem Downa, w tym trudności w uczeniu się, nieprawidłowości czaszkowo-twarzowe i hipotonia. Oprócz trudności w nauce, pacjenci z zespołem Downa borykają się z różnymi problemami zdrowotnymi, w tym z wrodzonymi wadami serca, chorobą Alzheimera (AD), białaczką, nowotworami i wadami przewodu pokarmowego. Zidentyfikowano 200-300 genów na chromosomie 21, które są odpowiedzialne za cechy kliniczne zespołu. Wiadomo, że wiele genów, takich jak polimorfizm cząsteczki adhezji komórkowej zespołu Downa (DSCAM) i genu APP, zarówno na chromosomie 21, jak i w innych regionach genomu, przyczynia się do zmienności objawów klinicznych.
Genetyka i typowe cechy zespołu Downa
Najczęstszą przyczyną urodzenia dziecka z zespołem Downa jest obecność dodatkowej kopii chromosomu 21, co skutkuje trisomią.
Trisomia 21 (47,XX,+ 21 lub 47,XY,+ 21) jest spowodowana brakiem rozdzielenia chromosomu 21 podczas rozwoju komórki jajowej lub plemnika. Innymi przyczynami mogą być translokacja Robertsonowska oraz chromosom izochromosomalny lub pierścieniowy.
Translokacja Robertsonowska występuje tylko w 2-4% przypadków i ma miejsce, gdy dłuższe ramię 21. chromosomu jest przyłączone do innego chromosomu submetacentrycznego. Mozaikowość powstaje w wyniku błędu w podziale komórki lub fałszywego podziału po zapłodnieniu. Dlatego u osób z mozaikowatością ZD w ich tkankach występują dwie linie komórkowe, z których jedna zawiera prawidłową liczbę chromosomów, a druga dodatkowy chromosom 21. Mozaikowość trisomii 21 i częściowa trisomia 21 są innymi rozpoznaniami genetycznymi i zwykle wiążą się z mniejszą liczbą cech klinicznych ZD. Mozaikowość trisomii 21 i częściowej trisomii 21 wiąże się na ogół z mniejszą liczbą cech klinicznych ZD.
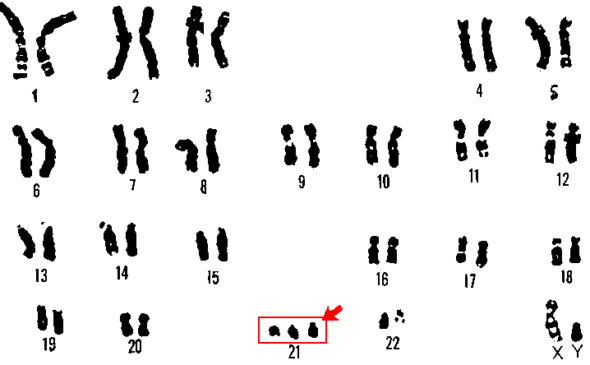
ZD charakteryzuje się dużą złożonością genetyczną i zmiennością fenotypu tj. zespołu cech organizmu. U osób z ZD występują pewne cechy fizyczne, takie jak mały podbródek, skośne oko, słabe napięcie mięśniowe, płaski mostek nosowy, pojedyncza bruzda na dłoni, duży palec u nogi, krótkie palce i duży język. U pacjentów z ZD może występować podwyższona wielkość mutacji lub liczba egzemplarzy, co może prowadzić do zwiększenia ekspresji genu Hsa 21. Określone geny, takie jak Hsa21 lub podzbiory genów, są w stanie kontrolować określone fenotypy ZD. Ponadto przeprowadzono analizy fenotypowe u osób z częściową trisomią dla Hsa21. Ustalono, że region o wielkości 3,8-6,5 Mb zwany “regionami krytycznymi zespołu Downa” (Down syndrome critical regions, DSCR) jest odpowiedzialny za większość fenotypów zespołu Downa w 21q21.22. Dzięki sekwencjonowaniu Hsa 21 uzyskano więcej informacji na temat korelacji genotyp-fenotyp w zespole Downa oraz charakterystyki regionów DSCR. Sugeruje się, że kinaza regulowana fosforylacją tyrozyny o podwójnej specyficzności (DYRK1A), regulator kalcyneuryny 1 (RCAN1) i cząsteczka adhezyjna komórek zespołu Downa (DSCAM) odgrywają kluczową rolę w rozwoju mózgu i występowaniu wad serca u pacjentów z ZD. W szczególności DSCAM odgrywa bardzo ważną rolę w różnicowaniu neuronów, prowadzeniu aksonów i tworzeniu sieci neuronalnych. Zaburzenia tych procesów przyczyniają się do powstawania anomalii neuropoznawczych w przebiegu ZD. Wszystkie badania wykazały, że nie ma jednego krytycznego obszaru genowego, który wystarczyłby do wywołania fenotypów ZD, natomiast musi istnieć duża liczba krytycznych miejsc lub krytycznych genów, które przyczyniają się do powstania fenotypu lub fenotypów związanych z ZD.
Różne rodzaje objawów związanych z zespołem Downa
Do różnych rodzajów zjawisk klinicznych związanych z zespołem Downa należą: choroba Alzheimera, wady serca, białaczka, nadciśnienie tętnicze i problemy żołądkowo-jelitowe. Mechanizm patogenezy tych fenotypów związanych z zespołem Downa powinien być badany wraz z czynnikami sprawczymi, aby lepiej zrozumieć tę chorobę.
Charakterystyka | Odsetek | Charakterystyka | Odsetek |
|---|---|---|---|
Upośledzenie umysłowe | 99% | Nieprawidłowe zęby | 60% |
Zachamowany wzrost | 90% | Skośne oczy | 60% |
Przepuklina pępkowa | 90% | Skrócone ręce | 60% |
Zwiększona skóra na karku | 80% | Krótka szyja | 60% |
Niskie napięcie mięśniowe | 80% | Obturacyjny bezdech senny | 60% |
Wąski podniebienie | 76% | Zagięty piąty palec | 57% |
Płaska głowa | 75% | Plamy zarośli w tęczówce | 56% |
Więzadła elastyczne | 75% | Pojedyncza poprzeczna fałda dłoniowa | 53% |
Proporcjonalnie duży język | 75% | Wystający język | 47% |
Nieprawidłowe uszy zewnętrzne | 70% | Wrodzona wada serca | 40% |
Spłaszczony nos | 68% | Zez | ~35% |
Oddzielenie pierwszego i drugiego palca | 68% | Niezstąpione jądra | 20% |
Choroba Alzheimera
Stwierdzono, że ryzyko wystąpienia choroby Alzheimera (Alzheimer Disease, AD) we wczesnym stadium jest wysokie u pacjentów z Zespołem Downa (ZD). Po 50tym roku życia u pacjentów z ZD ryzyko rozwoju otępienia wzrasta nawet o 70%. W ciągu ostatniej dekady dokonano znacznego postępu w poszukiwaniu genetycznych czynników ryzyka otępienia u osób z ZD oraz w zrozumieniu neuropatologicznych podobieństw i różnic między chorobą Alzheimera z ZD i bez niej. U osób z ZD w wieku powyżej 40 lat rozwój otępienia przebiega podobnie jak w przypadku choroby Alzheimera. Jeśli jednak otępienie występuje u osób młodszych (30-40 lat), objawia się zmianami osobowości i zachowania, takimi jak zwiększona impulsywność i początek apatii. Najbardziej widocznym podobieństwem między chorobą Alzheimera a AD z ZD są charakterystyczne neuropatologie, takie jak nagromadzenie amyloidu-β. Wyniki pośmiertnych badań neurochemicznych wykazały znaczny ubytek acetylotransferazy cholinowej i noradrenaliny u osób z ZD, co jest podobne do zmian obserwowanych w chorobie Alzheimera. Jak wynika z przeprowadzonych badań, dysregulacja układu cholinergicznego w przebiegu ZD jest kontrolowana przez gen DYRK1A. DYRK1A jest serynowo-treoninową kinazą białkową. DYRK1A jest zaangażowany w fosforylację białka tau, a jego podwyższenie może przyczyniać się do wczesnego tworzenia się splątków neurofibrylarnych. Ponadto wyniki uzyskane w badaniach mikromacierzy wskazują na wzrost regulacji podjednostki α2 i spadek regulacji podjednostek α3 i α5 receptora GABAA.
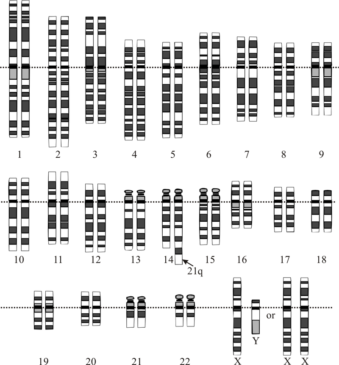
Istnieje kilka genów, o których wiadomo, że mogą powodować wczesny początek choroby Alzheimera. Najistotniejsze z tych genów to APP (amyloid precursor protein), BACE2 (Beta-secretase 2), PICALM (phosphatidylinositol binding clathrin assembly protein) i APOE (apolipoprotein E). APP jest integralnym białkiem błonowym koncentrującym się w synapsie neuronów. Uważa się, że trisomia tego białka może w znacznym stopniu przyczyniać się do zwiększonej częstości występowania otępienia u osób z ZD. Wykazano, że trisomia APP wraz z Hsa 21 u osób bez ZD wiąże się z wczesnym początkiem choroby Alzheimera. We wstępnym badaniu stwierdzono, że powtórzenie tetranukleotydowe ATTT w intronie 7 białka prekursorowego amyloidu wiąże się z wystąpieniem choroby u osób z zespołem Downa. Wiadomo również, że BACE2, kodujący enzym beta-sekretazę 2, odgrywa rolę w chorobie Alzheimera. Podobnie jak APP, gen BACE 2 jest zlokalizowany na chromosomie 21. Wyniki badań wskazują, że haplotypy w BACE2 są związane z chorobą Alzheimera. W badaniu obejmującym cały genom stwierdzono istotny związek między wariantami w BACE2 a wiekiem wystąpienia demencji w ZD, przy czym allel rs2252576-T wiązał się z wcześniejszym wystąpieniem demencji o 2-4 lata. W innych badaniach nie wykazano jednak istotnego związku między BACE2 a wiekiem wystąpienia demencji. Nadal nie ma pewności co do związku między wariantami BACE2 a rozwojem demencji w przebiegu ZD.
Oprócz genów APP i BACE2 stwierdzono, że inne geny, takie jak PICALM i APOE, są związane z wczesnym początkiem choroby Alzheimera w ZD. Zbadano PICALM, inny gen ryzyka dla AD i ZD. PICALM jest obecny w powiększonych endosomach we wczesnym stadium rozwoju AD. W badaniu obejmującym cały genom ZD potwierdzono związek między zmiennością w regionie PICALM na chromosomie 11 a wiekiem wystąpienia AD. Trzy występujące w tym badaniu warianty SNPS: rs2888903, rs7941541 i rs10751134 były związane z wcześniejszym wiekiem zachorowania na AD. Najważniejszym genetycznym czynnikiem ryzyka późnej postaci choroby Alzheimera jest allel ε4 genu APOE, znajdujący się na chromosomie 19. Allel APOE ε4, o którym wiadomo, że wiąże się ze zwiększonym obciążeniem amyloidowym i dysfunkcją układu cholinergicznego, jest prawdopodobnie najlepiej zbadanym genetycznym czynnikiem ryzyka. Wykazano, że u osób z ZD obecność allelu APOE ε4 zwiększa ryzyko wystąpienia choroby Alzheimera. Zwiększa się również akumulacja Aβ u osób z ZD będących nosicielami allelu APOE ε4.
Wady serca
Częstość występowania wad serca u noworodków z zespołem Downa wynosi do 50%. Wada zwana ubytkiem w poduszce przedsionkowo-komorowej jest najczęstszą wadą serca, występującą u 40% pacjentów z ZD. Ubytek przegrody międzykomorowej (VSD) również występuje u 35% pacjentów. W ubytku przegrody przedsionkowo-komorowej (AVSD) występuje wspólne połączenie przedsionkowo-komorowe w przeciwieństwie do prawidłowego serca. Inne wady to mięśniowe i błoniaste ubytki przegrody międzyprzedsionkowej oraz owalny kształt wspólnego połączenia przedsionkowo-komorowego. Tętnicze nadciśnienie płucne występuje u 1,2 do 5,2% osób z ZD. Wczesna terapia wad serca minimalizuje ryzyko wystąpienia niewydolności serca i nieodwracalnej choroby naczyń płucnych. Obserwacja specyficznych wzorców anatomicznych wad serca, które można zaobserwować w ZD, wykazała, że kluczową rolę w rozwoju wad serca odgrywa locus na chromosomie 21. Chociaż uważa się, że podwyższenie regulacji genów zmapowanych na chromosomie 21 jest związane z wadami serca, molekularne podstawy regulujące istnienie i anatomię wad serca są nadal niejasne. Sugeruje się, że kolagen typu VI (COL6A1, COL6A2) jest zaangażowany w patogenezę AVSD w zespole Downa, w podobny sposób jak inne geny zmapowane na chromosomie.
Poza chromosomem 21, inne geny zlokalizowane na różnych chromosomach również były badane jako przyczyna wad serca w ZD. Wśród tych genów gen CRELD1 został oceniony jako zwiększający podatność na AVSD. Stwierdzono, że mutacje w genie CRELD1 (Cysteine-rich EGF-like domain1) przyczyniają się do rozwoju AVSD w ZD. Gen CRELD1 jest zlokalizowany na chromosomie 3p25 i zawiera 11 eksonów o długości około 12 kb. Gen ten koduje białko powierzchni komórkowej, które pełni funkcję cząsteczki adhezji komórkowej i ulega ekspresji podczas rozwoju poduszeczek serca. Istnieją badania sugerujące, że gen CRELD1 prawdopodobnie odgrywa główną rolę w wywoływaniu fenotypu AVSD u osób z ZD. Dwie heterozygotyczne mutacje typu missense (p.R329C i p.E414K) zidentyfikowano u dwóch osób z ZD i AVSD. Do badania włączono również 39 osób z ZD z całkowitym AVSD i stwierdzono u nich te same mutacje. Nie wykryto takiej mutacji u osób z ZD bez wad serca. Mutację R329C opisano u osoby ze sporadycznym częściowym AVSD, wykryto ją również u osoby z ZD z AVSD. Chociaż mutacja jest taka sama u pacjentów z ZD, wada serca AVSD spowodowała poważniejszy stan. Dlatego sugeruje się, że mutacja CRELD 1 przyczynia się do patogenezy wad serca AVSD występujących u osób z ZD.
Nadciśnienie tętnicze
Osoby z ZD mogą mieć zwiększone ryzyko rozwoju nadciśnienia płucnego (PH), częściowo z powodu wrodzonych wad serca. Inne czynniki, takie jak niedrożność górnych dróg oddechowych, hipoplazja płuc z ZD, refluks żołądkowo-przełykowy, nieprawidłowa czynność naczyń płucnych mogą odgrywać rolę w zwiększaniu ryzyka rozwoju nadciśnienia płucnego w ZD. Wyniki badania przeprowadzonego w mieście Meksyk (na dużej wysokości) z udziałem chorych z ZD wykazały, że % z nich miało wrodzoną wadę serca, a 80% – zaburzenia funkcji płuc. Z drugiej strony, u osób z ZD odnotowano mniejszą częstość występowania nadciśnienia tętniczego.
Wykazano, że niektóre z miRs (klasa małych RNA) kodowanych przez Hsa21 ulegają nadekspresji w komórkach i tkankach pacjentów z zespołem Downa. Bezpośrednią przyczyną nadekspresji miRs w ZD wydaje się być dodatkowa kopia Hsa21, której miRs znajdują się w prawidłowej lokalizacji chromosomalnej. Stwierdzono, że trisomia Hsa21 mikroRNA hsa-miR-155 jest przyczyną tej niskiej częstości występowania. Specyficznym celem HsamiR-155 jest allel genu receptora angiotensyny II typu 1 (AGTR1). W badaniu bliźniąt (jedno z nich nie było zainfekowane, a drugie miało trisomię 21), mającym na celu ocenę ekspresji MiR-155 w trisomii 21, oba bliźnięta były homozygotyczne dla allelu 1166A AGTR1 i dlatego AGTR1 okazał się być celem miR-155. Receptor ten ma działanie wazopresorowe i reguluje wydzielanie aldosteronu. Jest to ważny czynnik kontrolujący ciśnienie i objętość krwi w układzie sercowo-naczyniowym. W związku z tym sugeruje się, że przyczynia się on do zmniejszenia ryzyka nadciśnienia tętniczego poprzez zmniejszenie ekspresji AGTR1. Potrzebne są dalsze badania, aby potwierdzić te przypuszczenia i ustalić, czy inne geny również mogą chronić osoby z zespołem ZD przed nadciśnieniem.
Białaczka
U pacjentów z ZD często występują nieprawidłowości hematologiczne. U chorych z zespołem Downa istnieje duże ryzyko wystąpienia nowotworów złośliwych, w tym białaczki. Pierwsze doniesienie o białaczce u pacjenta z ZD pochodzi z 1930 roku. Badania wykazały, że u pacjentów z ZD ryzyko wystąpienia białaczki jest około 10-20 razy większe, przy czym w wieku 5 lat wynosi 2%, a w wieku 30 lat – 2,7%. Osoby z ZD stanowią około 2% wszystkich przypadków ostrej białaczki limfoblastycznej u dzieci (ALL) i około 10% przypadków ostrej białaczki szpikowej (AML).
Mutacje somatyczne, takie jak genu GATA 1, odgrywają rolę w rozwoju ostrej białaczki megakarioblastycznej (AMKL) u pacjentów z ZD. GATA 1 jest czynnikiem transkrypcyjnym zlokalizowanym na chromosomie X, który odgrywa rolę w różnicowaniu erytroidów i megakariocytów. Mutacje w GATA 1 powodują krótszą ekspresję białka GATA 1 i w konsekwencji niekontrolowaną proliferację niedojrzałych megakariocytów. Przejściowa nieprawidłowa mielopoeza, forma preleukemii mieloidalnej, która występuje u około 10% noworodków z zespołem Downa, jest również spowodowana mutacjami w GATA1. Opisano, że mutacja w GATA1 u osób z ZD powoduje przemijające zaburzenia mieloproliferacyjne (TMD). Uważali oni, że jest prawdopodobne, iż trisomia 21 i GATA1 powodują hiperplazję wątroby płodu u niektórych osób z ZD, co może wywoływać okołoporodowe TMD.
Inną mutacją, której rolę sugeruje się w przypadkach ALL występujących w ZD, jest mutacja w genie Janus Kinazy 2 (JAK 2), występująca w około 30% przypadków ALL w ZD. Mutacje w szlaku JAK-STAT wiążą się z wysokim ryzykiem rozwoju ALL u osób z ZD. JAK2 jest niereceptorową kinazą tyrozynową i członkiem rodziny Janus Kinas. Bierze udział w sygnalizacji przez członków niektórych rodzin receptorów (np. receptorów interferonu i interleukiny). Mutacje w JAK2 są związane z polythemią vera, trombocytemią zasadniczą, mielofibrozą i innymi chorobami mieloproliferacyjnymi. Stwierdzono również, że geny JAK1, JAK2 i JAK3 są zmutowane u pacjentów z AMKL z ZD.
Wady przewodu pokarmowego
Osoby z zespołem Downa stanowią około 12% przypadków choroby Hirschprunga (HD). HD jest niedrożnością jelit spowodowaną brakiem prawidłowych komórek zwojów mięśniowych w części okrężnicy. W tej wadzie przewodu pokarmowego fale perystaltyczne nie przechodzą przez segment aganglionowy i powodują niedrożność, ponieważ nie dochodzi do normalnego wypróżnienia. Inne wady układu pokarmowego, które można zaobserwować u osób z ZD, to zwężenie dwunastnicy (DST) i niedomykalny odbyt (IA). Występują one odpowiednio 260 i 33 razy częściej w ZD. U noworodków z niedrożnością dwunastnicy lub DST we wczesnym okresie noworodkowym pojawiają się wymioty treścią pokarmową. Jeśli nie są leczone, istnieje ryzyko zgonu z powodu ciężkiego odwodnienia i zaburzeń równowagi elektrolitowej. Wada rozwojowa IA jest wadą wrodzoną, która powoduje zniekształcenie odbytnicy i wiąże się z częstszym występowaniem innych specyficznych anomalii, takich jak przetoka tchawiczo-przełykowa i atrezja przełyku.
Sugeruje się, że zmiany w genach niezwiązanych z Hsa21 odgrywają rolę w tych chorobach. Od dawna uważano, że to właśnie DSCAM jest potencjalnym czynnikiem tłumaczącym częstsze występowanie wad przewodu pokarmowego u pacjentów z zespołem Downa. DSCAM jest cząsteczką adhezyjną komórek zespołu Downa i odgrywa kluczową rolę w rozwoju ZD. Jest to białko transbłonowe, należące do nadrodziny immunoglobulin (Ig) – cząsteczek adhezji komórkowej. Ulega on ekspozycji w rozwijającym się układzie nerwowym, przy czym najwyższy poziom jego ekspozycji występuje w mózgu płodu. W przypadku nadmiernej ekspresji w rozwijającym się ośrodkowym układzie nerwowym płodu prowadzi do wystąpienia zespołu Downa. Gen DSCAM ulega ekspresji w grzebieniu neuronalnym, który daje początek jelitowemu układowi nerwowemu. Nakładający się region krytyczny jest zdefiniowany zarówno dla zespołu DST, jak i dla IA. Wykazano, że mutacje w genie DSCAM odgrywają rolę w rozwoju HD. W związku z rozwojem HD zidentyfikowano dwa SNPs, rs2837770 i rs8134673, obejmujące region wolny od eksonu 19 kb genu DSCAM.
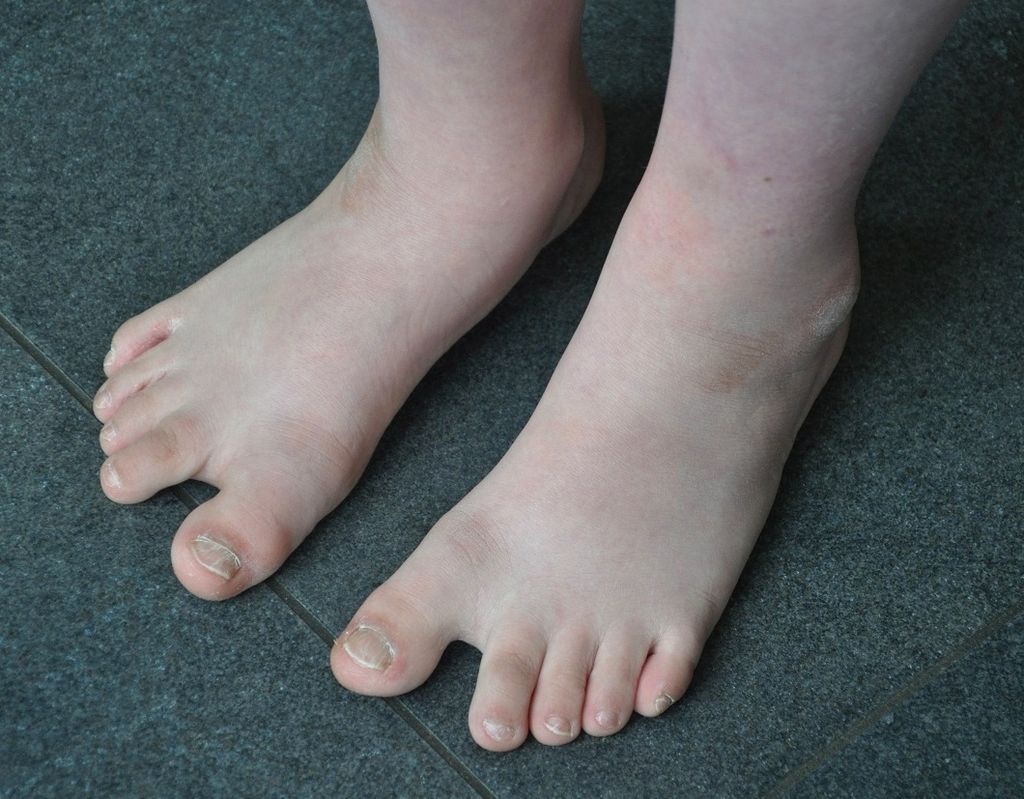
ZD, najczęstsza nieprawidłowość chromosomalna wśród noworodków, wiąże się z szeregiem wad wrodzonych, przede wszystkim z upośledzeniem umysłowym spowodowanym trisomią chromosomu 21. Oprócz własnych cech charakterystycznych, ZD mogą towarzyszyć różne rodzaje objawów fenotypowych. Aby zrozumieć interakcje między fenotypem a genotypem, rozważano różne teorie, takie jak “dawkowanie genów”. Fenotyp ZD jest spowodowany głównie brakiem równowagi w dawkowaniu genów zlokalizowanych na ludzkim chromosomie 21 (Hsa 21). Najczęstszą przyczyną ZD jest obecność dodatkowej kopii chromosomu 21. Uważa się, że krytyczny region w 21q22 jest odpowiedzialny za różne fenotypy ZD, takie jak nieprawidłowości twarzoczaszki, wrodzone wady serca, klinodaktylia i opóźnienie umysłowe. Problemy zdrowotne i długość życia osób z ZD są dość złożone i wiążą się z wieloma różnymi problemami medycznymi, psychologicznymi i społecznymi, występującymi od okresu niemowlęcego do dorosłości.
Związek ZD z różnymi fenotypami klinicznymi wymaga stałej obserwacji tych pacjentów z zastosowaniem podejścia multidyscyplinarnego. Na przykład, istnieją liczne badania epidemiologiczne i molekularne łączące zmiany patologiczne obserwowane w mózgach osób z zespołem Downa z neurodegeneracją obserwowaną w chorobie Alzheimera. Poznanie genów i patologii związanych z tymi zmianami jest bardzo ważne dla dobrej obserwacji klinicznej pacjentów z zespołem Downa. Ze względu na niewystarczającą wiedzę na temat molekularnej patogenezy ZD nie można na razie znaleźć skutecznej interwencji terapeutycznej. Sytuację dodatkowo komplikują złożone fenotypy towarzyszące ZD. Dobrym rozwiązaniem może być zastosowanie metod farmakologicznych w odniesieniu do kluczowych cząsteczek docelowych, które są kluczowe dla dysregulacji szlaków metabolicznych lub cech fenotypowych. Podsumowując, wyjaśnienie fenotypowych konsekwencji braku równowagi dawek genów w ZD oraz poznanie genów, które powodują towarzyszące im fenotypy, może stworzyć nowe możliwości interwencji terapeutycznych.
Źródło: Materiał na warunkach licencji Creative Commons 3.0, Autorzy:Fatma Söylemez




- Znakomity
- Bardzo Dobry
- Dobry
- Przeciętny
- Słaby
- Beznadziejny






Więcej
Czy ludzie agresywni powinni być izolowani i co się dzieje z ich mózgiem?
Czy Polsce grozi epidemia Ćpunów?
Ci ludzie są tak głupi a może tak mądrzy albo chorzy?